Genovar |
||||||||||
Kwang Su Jung1, Kiejung Park1, Sanghoon Moon2, Young Jin Kim2, Bong-jo Kim2 |
||||||||||
BackgroundAlong with single nucleotide polymorphisms (SNPs), copy number variation (CNV) is considered an important source of genetic variation associated with disease susceptibility. Despite the importance of CNV, the tools currently available for its analysis often produce false positive results due to limitations such as low resolution of array platforms, platform specificity, and the type of CNV. To resolve this problem, spurious signals must be separated from true signals by visual inspection. None of the previously reported CNV analysis tools support this function and the simultaneous visualization of comparative genomic hybridization arrays (aCGH) and sequence alignment. The purpose of the present study was to develop a useful program for the efficient detection and visualization of CNV regions that enables the manual exclusion of erroneous signals.
|
||||||||||
Screen shotsFigure 1. Chromosomal view and statistical summary .
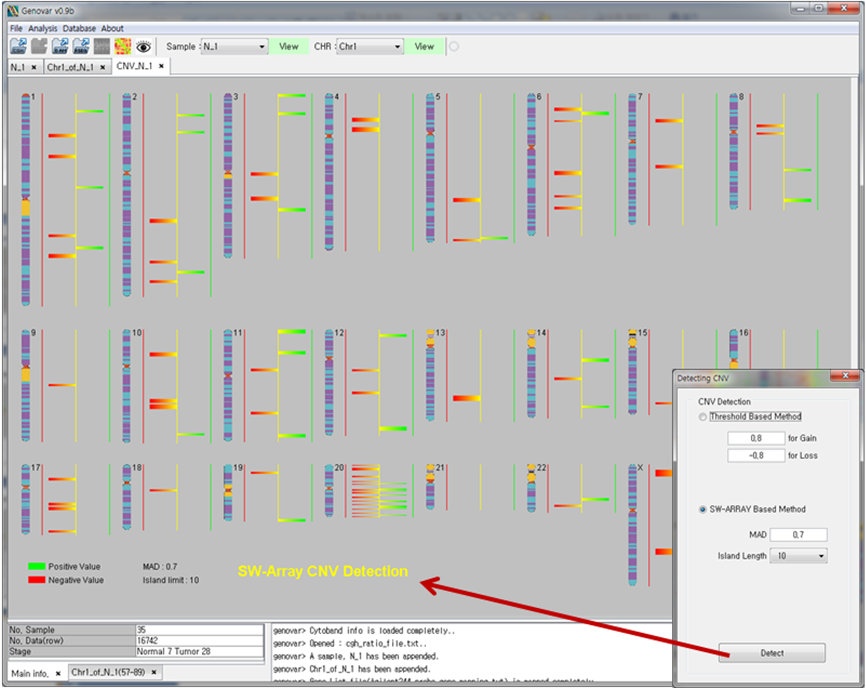
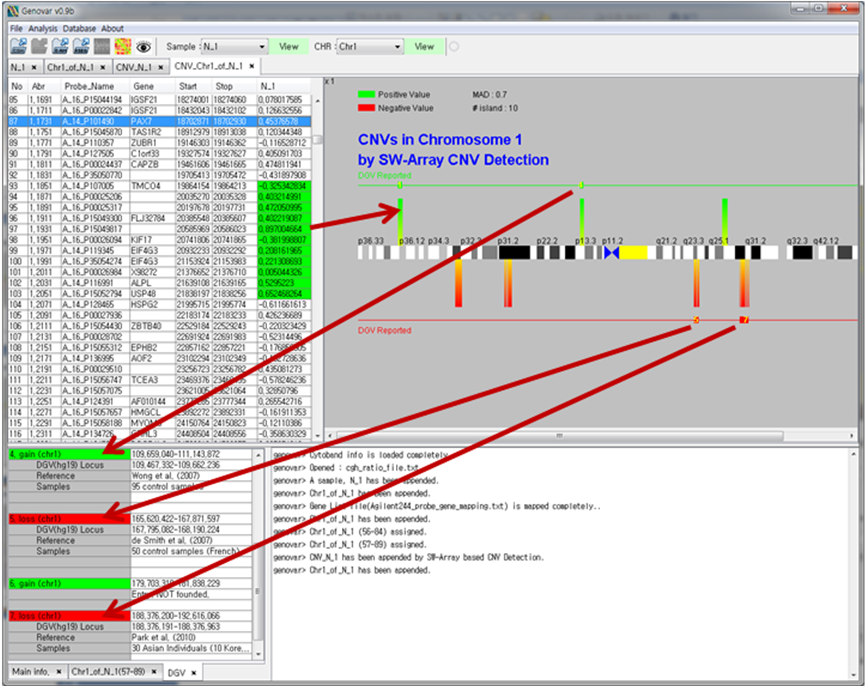
Figure 2. Copy number variation (CNV) detection.
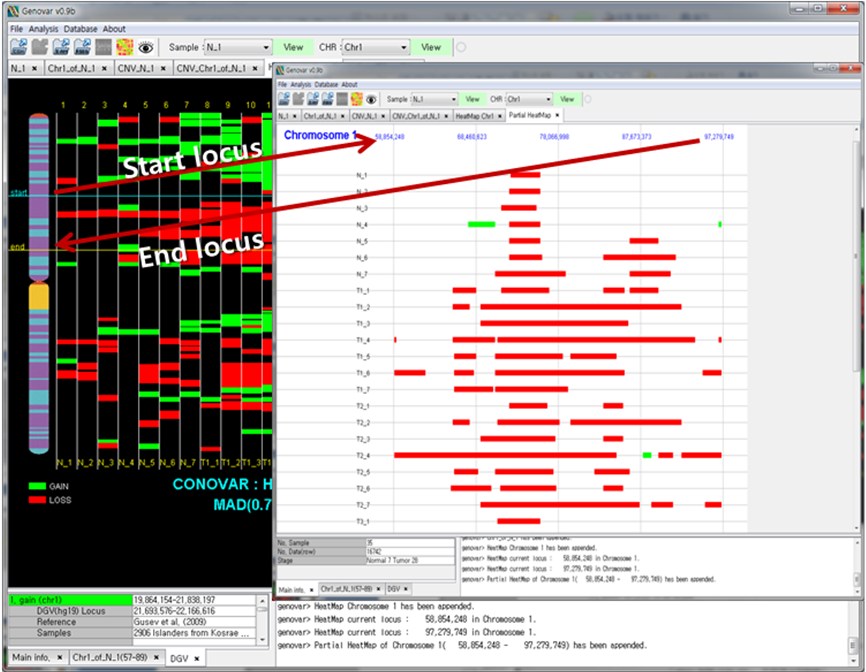
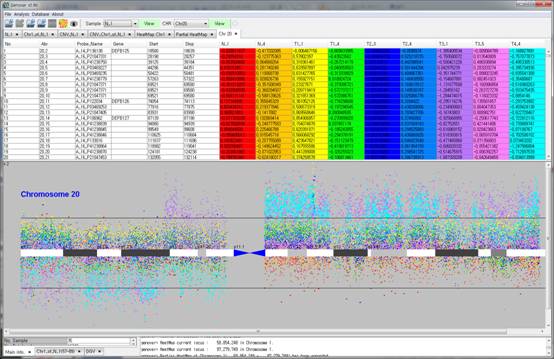
Figure 3. Comparison with multiple samples.
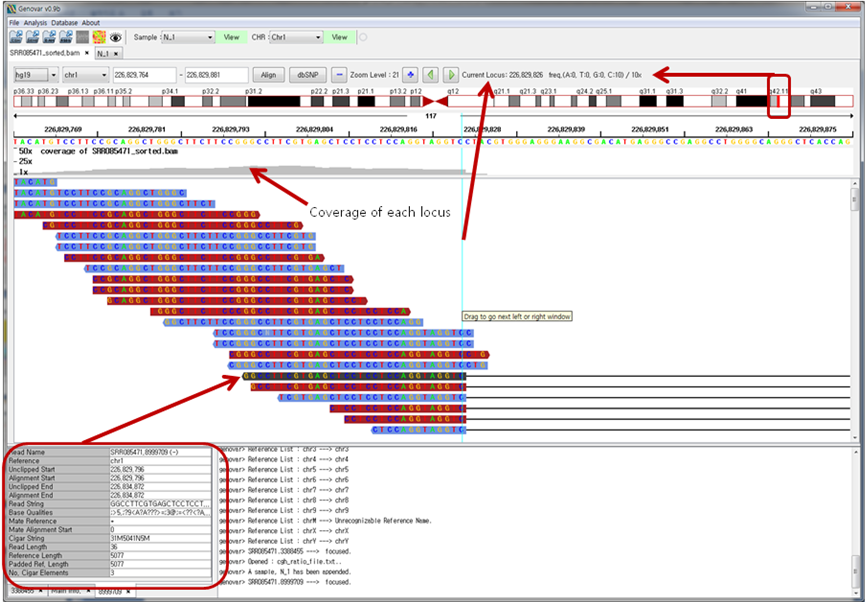
Figure 4. Sequence alignment view of a single binary sequence alignment/map (BAM) file.
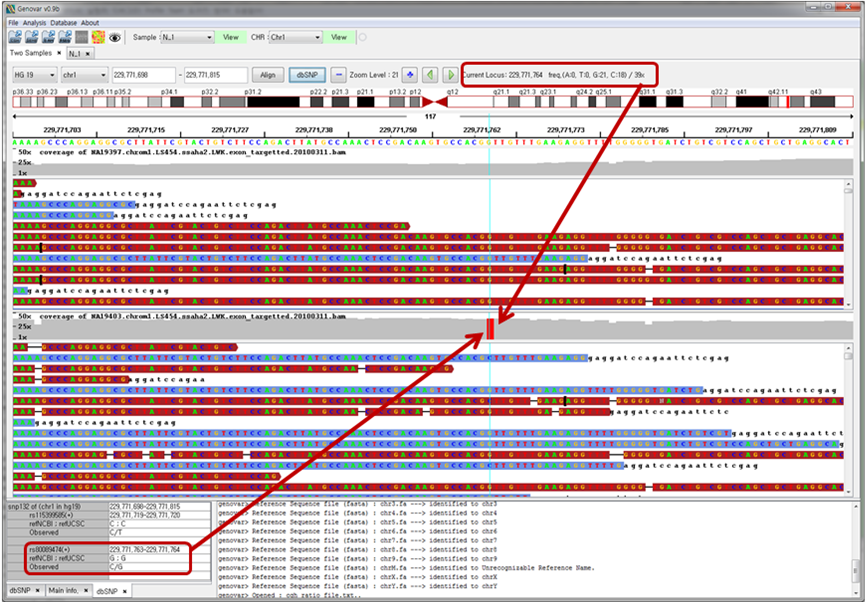
Figure 5. Sequence alignment comparison of two binary sequence alignment/map (BAM) files.
|